
神経難病について
筋萎縮性側索硬化症(Amyotrophic Lateral Sclerosis ALS)
ALSは筋肉を動かす運動神経細胞の変性により、全身の筋肉が徐々にやせて、力がなくなっていく病気です。中年以後に発症し、やや男性に多く、毎年人口10万人あたり約1名の新たな患者さんが発症しています。
原因
大部分の患者さんに遺伝性はなく、孤発性で原因は不明ですが、5~10%に家族性の方を認めます。
症状
手足の先から力が入りにくくなるタイプや、飲み込みづらさやしゃべりにくさなど、喉の症状から発症するタイプなどがありますが、様々な筋力低下・筋肉がやせる・ピクつきがでる、といった症状で始まります。いずれにせよ、やがて症状は呼吸筋も含めた全身に及びます。個人差はありますが、発症から3~5年で呼吸不全を呈します。
診断
脊椎疾患、末梢神経障害や筋疾患を除外した上で、経過を観察し診断に至ることが多い病気です。感覚障害、眼球運動障害、膀胱直腸障害、褥そうはALS陰性徴候といわれ、初期からこれらの症状が目立つ場合には他の疾患が考えられます。
治療
根治療法はなく、対症療法が主体となります。現在日本で使用できる薬剤は、進行を遅らせるとされるリルゾールのみで軽症の時期に服用を勧めます。リハビリテーションも大切な治療の一つですが、翌日にまで疲れが残るような運動は行いません。嚥下障害・呼吸筋障害などは生命予後に直結してくるため適切な時期に評価を行い、補助食品・経管栄養・補助呼吸装置などの方法を患者さんやご家族と一緒に考えていきます。症状の進行とともに言葉で自分の意思を伝えることが困難になってきますので、意思伝達装置の早めの導入を検討します。
パーキンソン病
パーキンソン病はふるえ・筋固縮・動作緩慢・小刻み歩行などを主症状とする病気で、人口10万人あたり100から150人の患者さんがいます。日本でも高齢化に伴い患者さんが増えています。
原因
中脳黒質の、ドパミンという神経伝達物質を産生する神経細胞が、徐々に障害され減っていくことが原因ですが、なぜ減るのかについてはまだよくわかっていません。通常遺伝性はありませんが、40歳以下で発症する若年発症の一部は家族性のこともあります。から150人の患者さんがいます。日本でも高齢化に伴い患者さんが増えています。
症状
1.安静時のふるえ 2.筋固縮 3.動作緩慢 4.姿勢反射障害 を四大症状とし、その他起立血圧、便秘、排尿障害などの自律神経障害も合併します。症状には左右差があることが殆どで、姿勢反射障害などは進行してから出現します。
診断
典型的な場合は、病歴・症状・L-DOPA製剤が有効という点から診断可能ですが、他のパーキンソン症状を示す症候群との鑑別が困難な場合もあります
治療
治療の基本は薬物治療です。不足しているドパミンを補充する、L-DOPA製剤、ドパミン受容体を刺激するアゴニスト、古くからある抗コリン剤、塩酸アマンタジン、すくみ足を改善するドロキシドパ、MAO-B阻害薬、末梢性COMT阻害薬など多くの薬剤があり、症状に応じて組み合わせて使用します。手術療法は定位脳手術によって行われる脳深部刺激療法ですが、年齢や症状によって適応を検討します。脳神経外科のサイトもご参照ください。
脊髄小脳変性症(Spino-Cerebellar Degeneration SCD)
小脳、脳幹や脊髄に障害をきたし、小脳性運動失調を主症状とする、神経変性疾患の一群の総称です。“変性”とは、原因がわからないものの、神経細胞が徐々に障害されて死滅していくことを示します。我が国の人口10万人あたり5-10人の患者さんがいるとされています。
原因
遺伝性が約30%、孤発性が約70%で、孤発性の多くは現在は多系統萎縮症とされているオリーブ橋小脳萎縮症の方で、そのほかに皮質性小脳変性症があります。遺伝性脊髄小脳変性症のうち、多くは常染色体優性遺伝形式をとり、これらは原因となる遺伝子により脊髄小脳失調症1型、2型などと分類されます。そのうち、日本では、脊髄小脳失調症3型(Machado-Joseph病=マシャド・ジョセフ病)の方が約27%と最も多く、ほかに脊髄小脳失調症6型(約21%)や歯状核赤核淡蒼球ルイ体萎縮症(DRPLA)(約10%)の方もいらっしゃいます。
症状
症状の中心は小脳性運動失調です。これは、歩行時や起立時にふらつく、歩幅を広くとり、あたかも酔ったような歩き方になる、動作時の手の震え、細かな動作が下手になる、呂律がまわりにくくなる、などの症状として現れます。そのほかには、筋肉のつっぱりや排尿障害、不随意運動がみられる場合もあります。症状や進行する速度は、病型により異なりますし、さらに個人によって異なります。
診断
まず、臨床症状と経過、家族歴が大事です。さらに頭部MRI検査などを行います。また、原因となる遺伝子異常がわかっている場合には、御相談の上、遺伝子解析検査を行う場合もあります。
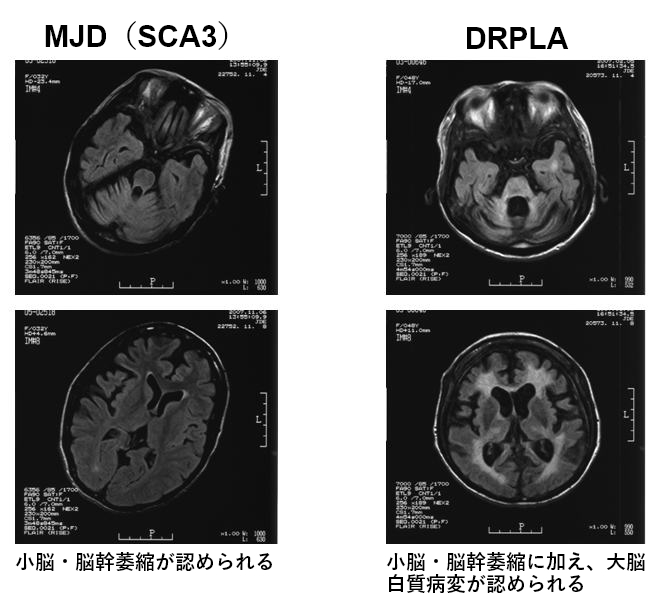
治療
現在の医学では、根治させる治療法はありませんが、病気が進行するのを防ぐために、トリチレリン水和物(セレジスト®)の内服治療を行います。このほか、個々の症状に応じた治療を行います。さらに、日常生活を送る上で不自由をより少なくし、病気の進行による機能の低下を防ぐために、リハビリテーションを継続することが大事です。また、自宅でより安全に過ごしやすくなるように、段差をなくすなど、環境を整えることも大事です。
多系統萎縮症(Multiple System Atrophy MSA)
かつて、線条体黒質変性症(SND)・オリーブ橋小脳萎縮症(OPCA)・シャイ・ドレーガー症候群(SDS)として別々に診断されていた3つの疾患を、病理学的に同じ系統が障害されることから統一したものです。人口10万に数人程度の患者さんがいます。
原因
遺伝性はありません。
症状
SNDではパーキンソン症状が、OPCAでは小脳失調症状が、SDSでは自律神経症状がそれぞれ初発症状で前景に出ることが普通ですが、長い経過中には最初の症状が目立たなくなり、数年~10年以内には寝たきりとなります。パーキンソン症状に対して、病初期に抗パーキンソン病薬が有効なことがありますが、パーキンソン病とは異なり通常その効果は一過性です。また、MSAの患者さんでは突然死することがあり、そのひとつの原因として声帯開大不全による睡眠時呼吸障害があります。これは夜間睡眠時に吸気時の喘鳴として認められるもので終夜ポリグラフによる評価を行い、必要に応じ睡眠時の補助呼吸などの導入を図ります。
診断
病歴、臨床症状の経過、薬剤の効果などから診断します。頭部MRI検査でOPCAであれば橋底部に見られる十字サインと呼ばれる白質の変性所見が、SNDであれば被核外側の線状T2高信号域が、診断の際に参考になります。
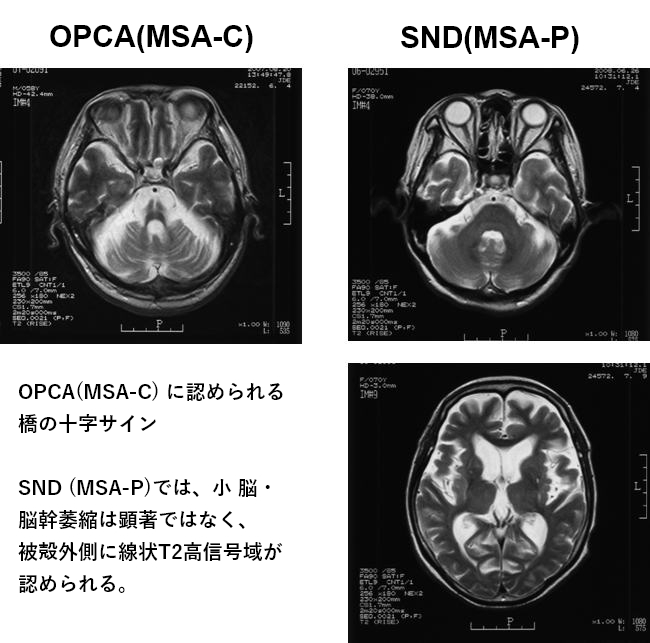
治療
根治療法はなく、対症療法が主体です。パーキンソン症状に対してはパーキンソン病治療薬を充分投与してみることが大切です。起立性低血圧はSDSでは特に重症なことがあり、そのために寝たきりとなる場合があります。充分に対処しないまま移動時に失神して転倒し骨折することがあり注意が必要です。また機能維持のためリハビリテーションを継続することも大事です。
神経免疫疾患 (多発性硬化症・慢性炎症性脱髄性多発神経根炎(CIDP)・重症筋無力症など)
免疫とは、本来、細菌などの外敵から身を守るための機能ですが、それが自分に向けられた状態を自己免疫疾患と総称します。神経内科領域においても多発性硬化症、慢性炎症性脱髄性多発根神経炎、重症筋無力症などがその範疇に入ります。
多発性硬化症は中枢神経線維を覆う髄鞘が壊される病気です。症状は、障害される部位により視力障害から運動麻痺、感覚障害、排尿障害などさまざまです。治療は、急性期にはステロイドの治療(点滴→内服)、慢性期にはリハビリテーションや対症療法、再発予防にはインターフェロンβを行います。多くは再発・寛解を繰り返し、一部は次第に進行性の経過をとります。
慢性炎症性脱髄性多発根神経炎(CIDP)は末梢神経線維を覆う髄鞘が壊される病気です。手足の感覚障害や運動麻痺などをきたします。治療はステロイド療法、免疫グロブリン静注療法、血漿交換療法、免疫抑制療法などがあります。
重症筋無力症は筋肉と末梢神経の接合部が障害される病気です.症状は筋肉の易疲労性や筋力低下です.眼に限局するタイプと全身に症状が及ぶタイプがあります.治療は対症療法としてのコリンエステラーゼ阻害薬の内服や,免疫療法として胸腺摘出術やステロイドや免疫抑制剤の内服,血漿交換療法などがあります.治療により約半数の方が発症前と同じ状態に回復します。